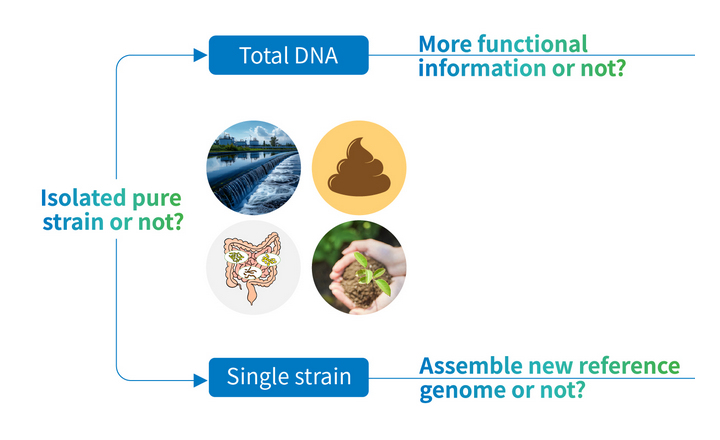
- Shallow metagenomics is a low-depth implementation of shotgun metagenomic sequencing proposed as an alternative to both 16S rRNA gene amplicon sequencing (limited taxonomic resolution and amplification bias) and deep shotgun sequencing (high cost). It typically involves sequencing ≤1 Gb (~3 million reads). Accordingly, it is increasingly being applied to microbiome research and surveillance is increasing, but the accuracy limits of strain inference and genome reconstruction under low-depth conditions remain incompletely defined.
- Metagenome-assembled genomes (MAGs) are widely used to reconstruct microbial genomes directly from metagenomic sequencing data and are increasingly applied to strain-level analysis, transmission inference, and functional characterization. However, reliable MAG reconstruction requires sufficient genome coverage, making MAG quality highly dependent on sequencing depth and assembly performance.
- Because many downstream microbiome studies rely on MAGs, establishing practical sequencing-depth thresholds is essential for designing reliable metagenomic studies and accurately interpreting transmission, persistence, functional, and ecological signals.
Key Findings: To better define the strengths and limitations of shallow metagenomics and provide guidance for future studies, the authors systematically evaluated the effects of shotgun sequencing depth on taxonomic composition, strain-level diversity, and functional inference using complex bacterial mock communities sequenced at depths ranging from 0.1–50 Gb.¹ Three mock microbial communities were constructed, libraries were generated using the Nextera XT DNA Library Preparation Kit (Illumina), and sequencing was performed on an Illumina NovaSeq 6000 platform.
- Reference-based taxonomic profiling at shallow depth: Reliable taxonomic and strain-level profiling at ~1–5 Gb was achieved when high-quality reference genomes were available. However, performance was strongly abundance-dependent. Low-abundance genomes (0.00046–0.0046% relative abundance) achieved only 0.8–21.8% genome coverage even at the highest sequencing depth (50 Gb), whereas the most abundant strains (>4.6% relative abundance) achieved 40.5–98.3% coverage at only 0.1 Gb. Although all strains were detectable at 0.1 Gb, most genomes (63–91%) exhibited low overall coverage (0–25%) at this depth.
- Strain-level discrimination exceeded MetaPhlAn performance: Using reference genomes, closely related strains with very high average nucleotide identity (ANI) could still be reliably distinguished at the strain level. In contrast, MetaPhlAn, which relies on marker-gene profiling rather than whole-genome resolution, showed increased deviation from expected relative abundances and frequently collapsed within-species strain diversity into a single signal. These discrepancies were most pronounced at lower sequencing depths and in more complex mock communities, reflecting reduced accuracy under shallow sequencing conditions and limited strain-level resolution.
- Chimeric MAGs inflated apparent strain diversity: De novo metagenome-assembled genome (MAG) reconstruction required >10 Gb sequencing depth and remained prone to fragmentation and chimerism even among MAGs classified as “high quality” based on standard completeness and contamination metrics. Depending on the workflow, only 54.5–81.8% of MAGs accurately reconstructed the original strains, with remaining cases reflecting strain splitting, strain merging, or chimeric assemblies rather than true genome recovery.
- Long-read sequencing and strain-aware assembly improved MAG quality: Multicoverage binning, strain-aware assembly (metaSPAdes), and long-read sequencing reduced—but did not eliminate—MAG chimerism by improving separation of closely related strains and increasing assembly contiguity. Among the evaluated approaches, long-read sequencing produced the highest proportion of coherent MAGs, although residual strain mixing and assembly ambiguity persisted.
- Functional inference depended on sequencing depth: pathway-level functional profiling remained relatively stable at ~2–5 Gb due to redundancy in functional annotation, whereas comprehensive gene- and protein-level recovery required substantially deeper sequencing (≥10 Gb), particularly in complex communities where low-abundance functions were under-sampled at shallow depth.
- Wet-lab and host-DNA effects were important confounders: Library preparation methods and host/background DNA contamination significantly affected taxonomic and functional performance at shallow sequencing depths (<5 Gb) by altering the proportion of informative microbial reads and introducing systematic representation bias. At low sequencing depth, even modest increases in host or non-target DNA effectively reduce microbial coverage, disproportionately impacting detection of low-abundance taxa, strain-level resolution, and functional gene recovery. Differences in library preparation further compound these effects through variability in DNA fragmentation, amplification bias, and uneven representation of genomic regions, ultimately influenced both apparent community composition and inferred functional profiles.
Bigger Picture: This work clarifies a key methodological boundary in metagenomics: increasing sequencing depth improves resolution up to a point, after which returns diminish for reference-based analyses but remain necessary for de novo genome reconstruction. It underscores that sequencing depth–dependent recovery is strongly influenced by host/background DNA contamination, library preparation method, and microbial community complexity, which together influence the effective microbial read depth and detection sensitivity. Strain abundance and genome complexity further affect recovery accuracy and completeness. Shallow metagenomics is suitable for broad taxonomic and pathway-level functional profiling but is insufficient for accurate de novo strain reconstruction, which requires deeper sequencing and remains constrained by MAG assembly artifacts and strain-level ambiguity.
(Image Credit: iStock/JuSun)